Definition and Genetic Mechanism
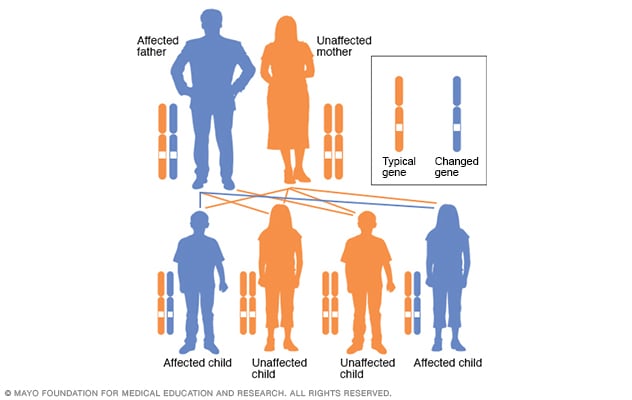
Huntington’s disease (HD) is a progressive neurodegenerative disorder caused by an abnormal expansion of CAG trinucleotide repeats in the HTT gene on chromosome 4, leading to toxic protein accumulation that damages brain cells over time. This autosomal dominant condition follows a clear inheritance pattern where each child of an affected parent has a 50% risk of inheriting the mutated gene, with the number of CAG repeats directly determining disease severity and age of onset—typically 40+ repeats for adult-onset (30s-40s) and 60+ for juvenile forms. As the mutated gene passes through generations, a phenomenon called genetic anticipation may cause earlier symptom onset due to further repeat expansion, particularly with paternal inheritance, though individual symptom progression can vary significantly. While facing this diagnosis can be overwhelming, genetic counseling through organizations like the Huntington’s Disease Society of America provides crucial support for understanding risks and making informed family planning decisions.
Primary Motor Symptoms
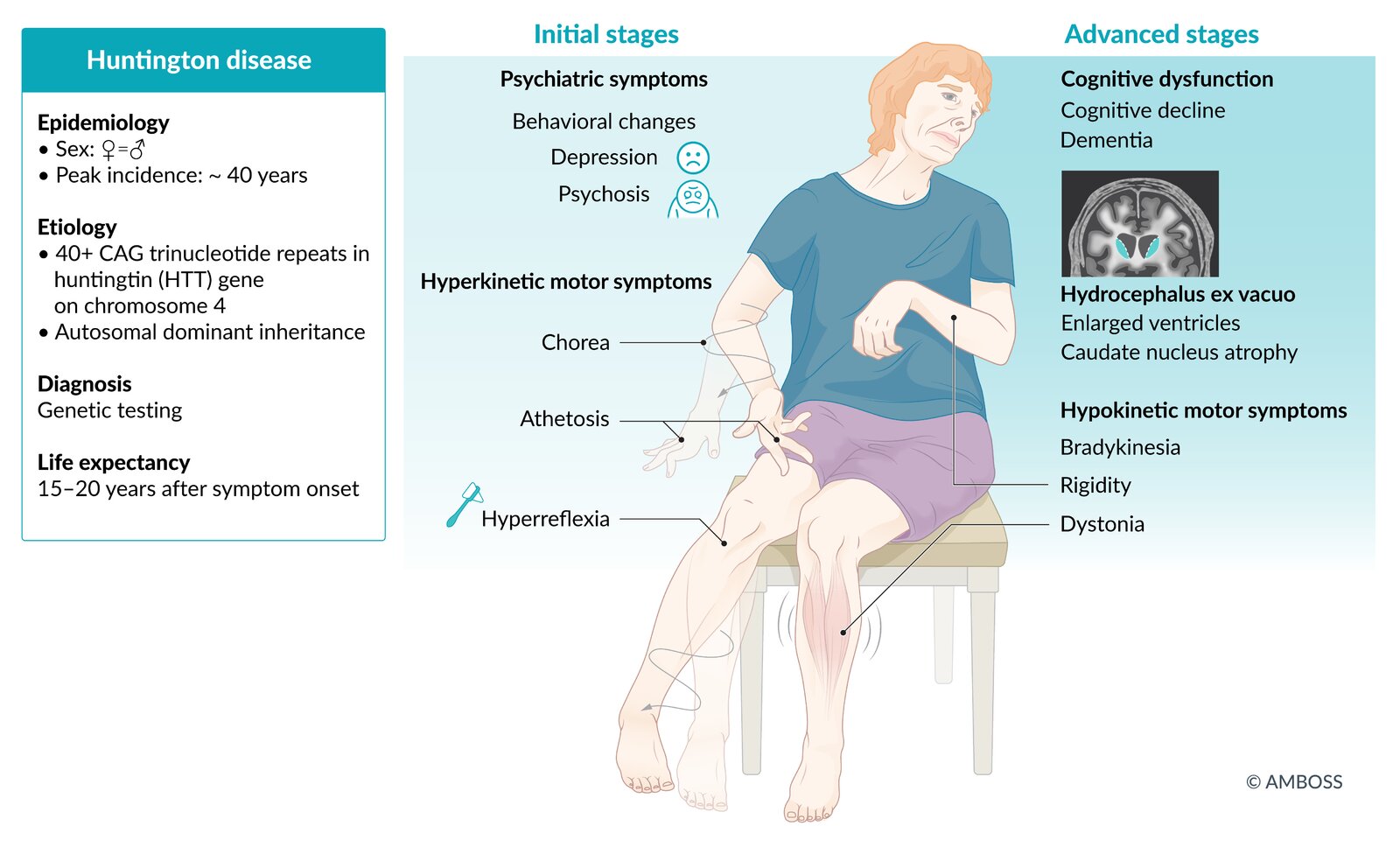
Chorea—the hallmark involuntary jerking movement—typically emerges first as subtle finger tapping or facial twitching before evolving into pronounced, dance-like motions that disrupt daily function. Per University of Utah Health research, 85-90% of adult-onset patients experience this uncontrolled hyperkinetic movement, which often progresses from mild limb fidgeting to whole-body spasms within 5-7 years. Balance challenges become increasingly apparent as gait instability leads to frequent stumbling, while muscle rigidity in juvenile cases (onset <20 years) creates Parkinsonian slowness rather than chorea. Speech gradually deteriorates from mild slurring to near-unintelligibility, and swallowing difficulties may necessitate dietary modifications to prevent aspiration—yet consistent physical therapy at specialized centers like the HDSA Centers of Excellence can significantly prolong functional mobility and safety.
Cognitive Decline Indicators
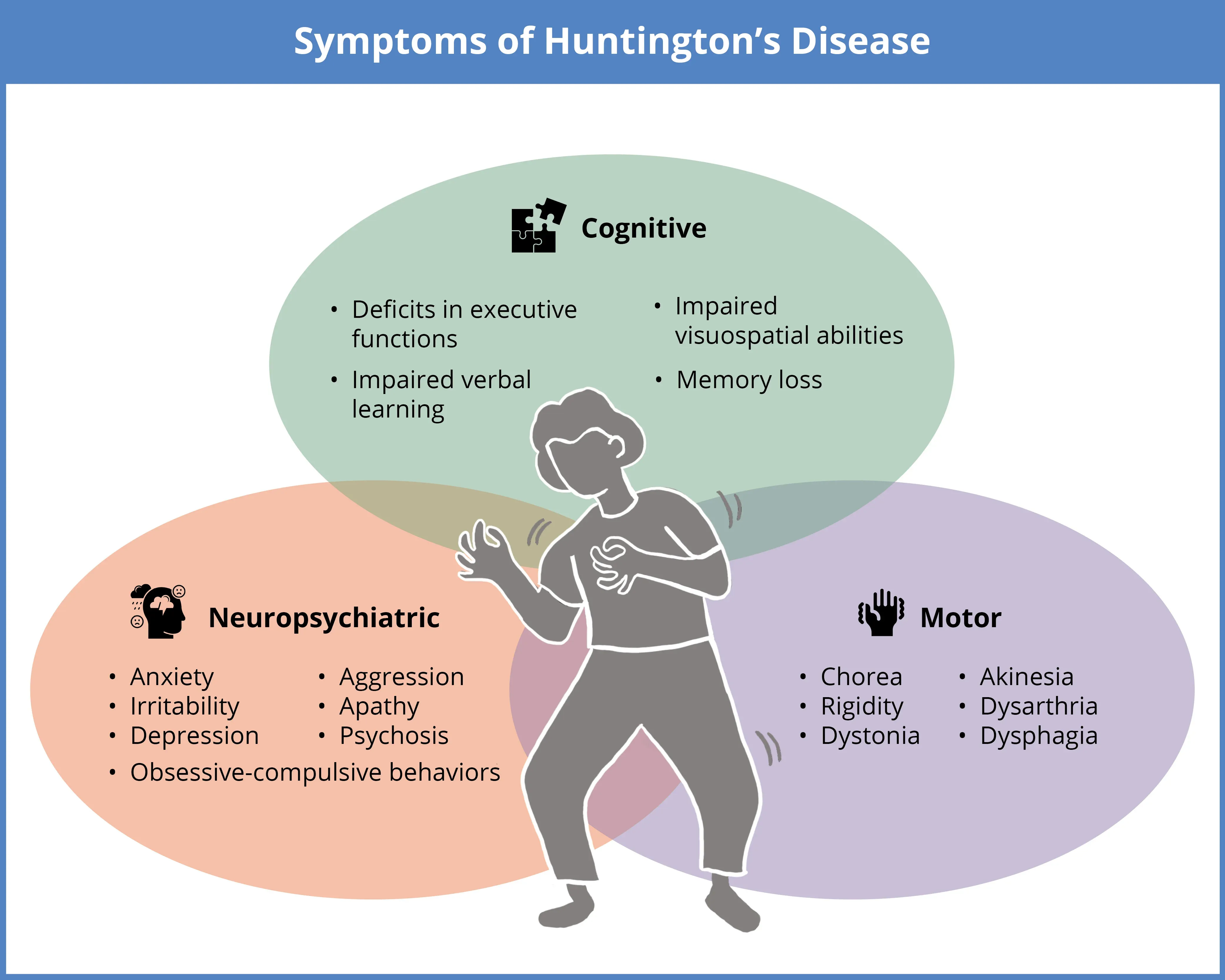
Early cognitive changes in Huntington’s disease typically begin with subtle executive function impairments—difficulty concentrating, poor judgment, and challenges with planning—often preceding noticeable memory loss. According to JAMA Neurology research, 70% of patients experience slowed processing within 3-5 years of motor symptom onset, making complex tasks like budgeting or following conversations increasingly difficult. Unlike normal age-related forgetfulness where knowledge remains intact, HD progressively erodes the ability to learn new information and safely manage daily activities, though cognitive therapy at HDSA Centers of Excellence can significantly extend functional capacity. Recognizing early indicators enables timely implementation of practical strategies like memory aids, simplified routines, and occupational therapy to maintain independence during disease progression.
Psychological and Emotional Changes

Psychological symptoms frequently emerge before motor manifestations in Huntington’s disease, with depression affecting approximately 50% of patients and anxiety impacting over 30% according to the Huntington’s Disease Society of America. Individuals commonly experience dramatic mood fluctuations, irritability, emotional blunting, and obsessive-compulsive tendencies that strain personal relationships, while some develop more severe psychiatric symptoms like delusions or hallucinations as the disease advances. These emotional transformations result from progressive neurodegeneration in mood-regulating brain circuits rather than personal failings, making compassionate understanding essential for families navigating these difficult changes. Practical approaches including structured daily routines, professional counseling through HDSA support networks, and participation in caregiver support groups can significantly alleviate emotional distress and foster resilience throughout the disease journey.
Diagnostic Testing Methods
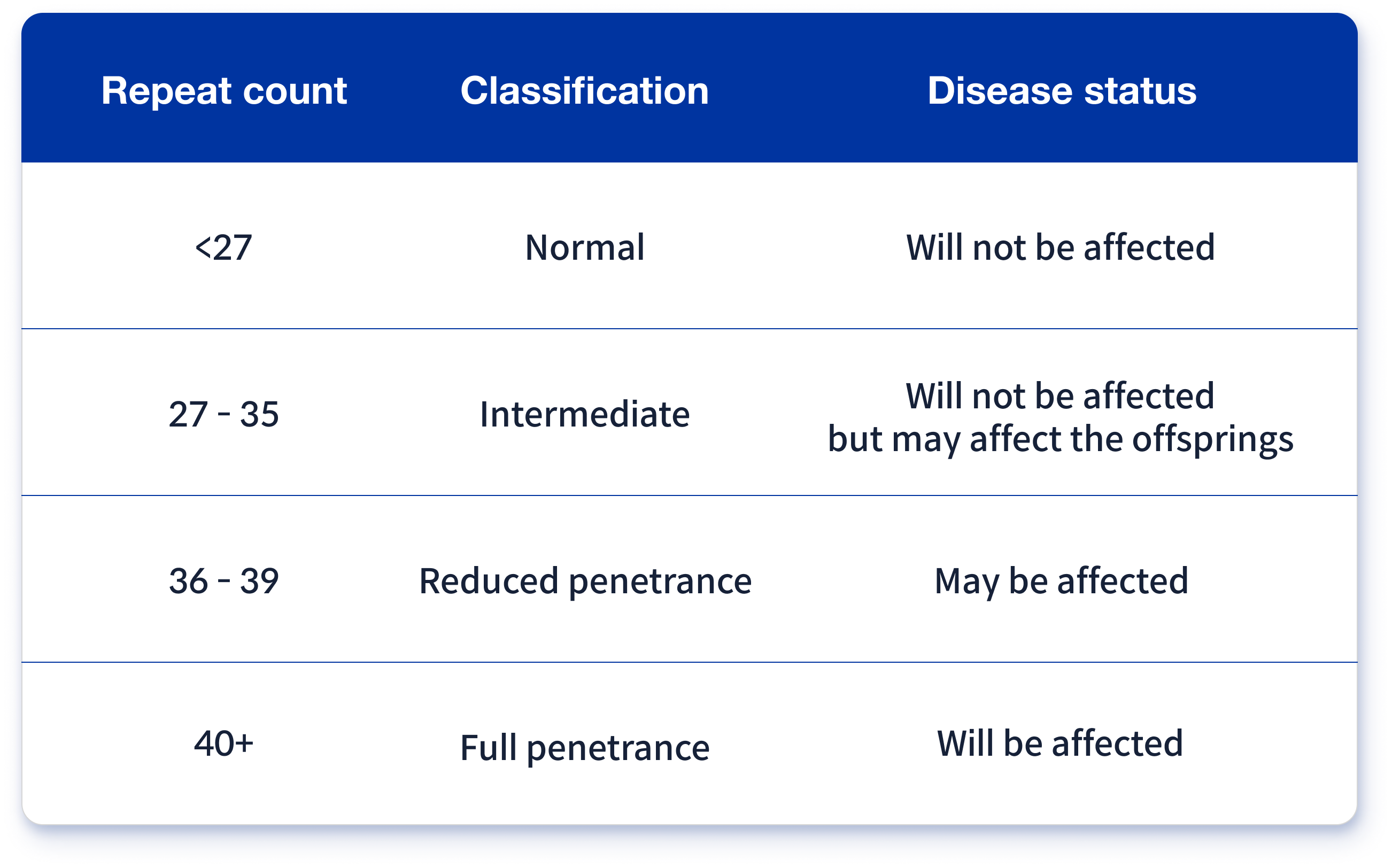
Accurate diagnosis of Huntington’s disease follows a systematic process beginning with thorough clinical evaluation at specialized centers like the Huntington’s Disease Society of America (HDSA) Centers of Excellence. Neurologists utilize the Unified Huntington’s Disease Rating Scale (UHDRS) to objectively assess motor function through standardized tasks measuring chorea severity, balance, and coordination with specific metrics like finger-tapping speed and gait stability. Cognitive screening with tools such as the Symbol Digit Modalities Test identifies early executive function changes, while brain imaging (MRI/CT) helps exclude other neurological conditions and may reveal characteristic caudate nucleus atrophy in later stages. Definitive diagnosis requires genetic testing that quantifies CAG trinucleotide repeats in the HTT gene, where 40+ repeats confirms Huntington’s disease—though comprehensive genetic counseling before and after testing through organizations like the National Society of Genetic Counselors provides essential emotional support and helps individuals navigate complex decisions with compassion.
Symptom Management Strategies

Though Huntington’s disease currently has no cure, proactive symptom management through HDSA Centers of Excellence significantly enhances daily living quality across all disease stages. For chorea (involuntary jerking movements), FDA-approved medications like tetrabenazine regulate dopamine to reduce symptom severity, while physical therapy with balance exercises and adaptive tools such as weighted utensils maintains functional independence longer. Cognitive strategies including memory notebooks, simplified routines, and occupational therapy sessions help preserve executive function, and antidepressants combined with HDSA counseling effectively address the 50% depression rate common in HD patients. A multidisciplinary care team—integrating neurologists, psychiatrists, and genetic counselors—ensures personalized care that adapts to evolving symptoms, with recent clinical trials of gene-silencing therapies offering new hope for slowing disease progression.